


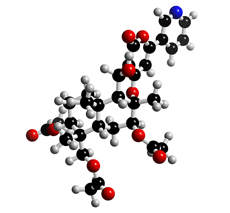
The molecular structure of Pyripyropene A
Click on the image above to interact with the 3D model of the Pyripyropene A structure
Pyripyropene
Discovery, producing organism and structures (1-6, 16-18)
Pyripyropenes were isolated from a culture broth of the fungal strain FO-1289 and found by an enzyme assay to be inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). The structures were elucidated by spectral analysis, including NMR (3,5). The relative and absolute stereochemistries were confirmed by NOE and X-ray crystallographic analysis for pyripyropenes A (4) and E (5). Pyripyropenes have a common structure consisting of pyridine, α-pyrone and sesquiterpene moieties.
Incorporation of 13C-labeled precursors and degradation experiments revealed that a nicotinic acid primer condenses with two acetates in a head-to-tail fashion forming the pyridino-α-pyrone moiety, which is linked with a sesquiterpene forming the core skeleton. Then, three acetyl residues are introduced into the core. This is the first demonstration that an intact nicotinic acid works as an acyl primer unit for oligoketide formation in fungal secondary metabolites. The total syntheses of pyripyropenes have been reported by several groups.
Physical data (Pyripyropene A) (3)
White powder. C31H37NO10; mol wt 583.24. Sol. in MeOH, EtOAc, CHCl3. Insol. in H2O, hexane.
Biological activity (1,2,4, 7–15)
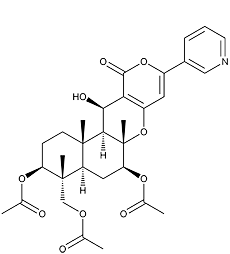
Pyripyropene A
1) Enzyme assay for ACAT inhibition (1,2,7)
ACAT inhibitory activity was tested in an enzyme assay using rat liver
microsomes. Pyripyropenes are the most potent ACAT inhibitors of microbial origin.
2) Structure-activity relationship (8-12)
Comparison of ACAT inhibitory activity among natural pyripyropenes and synthetic derivatives (8–12) revealed the structure-activity relationship.
In vivo efficacy of pyripyropene A and synthetic derivatives were evaluated in a hamster model. PR-86 showed more potent inhibition of cholesterol absorption from the intestines (ED50, 10 mg/kg) than pyripyropene A (ED50, about 100 mg/kg).
4. Inhibition of multidrug resistant P-glycoprotein by a semi-synthetic analog of pyripyropene. (14,15)
7-O-Benzoylpyripyropene (7-O-BzP) (6.25 µg/ml) effectively reverses P-glycoprotein-related
MDR by interacting directly with P-glycoproteins in drug resistant VJ-300 and
P388/ADR cells.
References
1. [508] S. Ōmura et al., J. Antibiot., 46, 1168–1169 (1993)
2. [534] H. Tomoda et al., J. Antibiot., 47, 148–153 (1994)
3. [535] Y. K. Kim et al., J. Antibiot., 47, 154–162 (1994)
4. [559] H. Tomoda et al., J. Am. Chem. Soc., 116, 12097–12098 (1994)
5. [574] H. Tomoda et al., J. Antibiot., 48, 495–503 (1995)
6. [609] H. Tomoda et al., J. Antibiot., 49, 292–298 (1996)
7. A. S. Katocs et al., FASEB J., 2, A 1219 (1988)
8. [585] R. Obata et al., J. Antibiot., 48, 749–750 (1995)
9. [596] R. Obata et al., Bioorg. Med. Chem. Lett., 5, 2683–2688 (1996)
10. [644] R. Obata et al., J. Antibiot., 49, 1133–1148 (1996)
11. [645] R. Obata et al., J. Antibiot., 49, 1149–1156 (1996)
12. [657] R. Obata et al., J. Antibiot., 50, 229–236 (1997)
13. [689] R. Obata et al., Chem. Lett., 935–936 (1997)
14. [748] R. Obata et al., J. Antibiot., 53, 422-425 (2000)
15. [761] M.-C. Rho et al., J. Antibiot., 53, 1202-1206 (2000).
16. [604] H. Tomoda et al., J. Org. Chem., 61, 882–886 (1996)
17. [600] T. Nagamitsu et al., J. Org. Chem., 60, 8126–8127 (1995)
18. [637] A. B. Smith, III et al., Tetrahedron. Lett., 37, 6461–6464 (1996)